PRISTIQ® Clinical Studies
(desvenlafaxine succinate)
14 CLINICAL STUDIES
Major Depressive Disorder
The efficacy of PRISTIQ as a treatment for depression was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose studies (at doses of 50 mg per day to 400 mg per day) in adult outpatients who met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for major depressive disorder. In the first study, patients received 100 mg (n = 114), 200 mg (n = 116), or 400 mg (n = 113) of PRISTIQ once daily, or placebo (n = 118). In a second study, patients received either 200 mg (n = 121) or 400 mg (n = 124) of PRISTIQ once daily, or placebo (n = 124). In two additional studies, patients received 50 mg (n = 150 and n = 164) or 100 mg (n = 147 and n = 158) of PRISTIQ once daily, or placebo (n = 150 and n = 161).
PRISTIQ showed superiority over placebo as measured by improvement in the 17-item Hamilton Rating Scale for Depression (HAM-D17) total score in four studies and overall improvement, as measured by the Clinical Global Impressions Scale - Improvement (CGI-I), in three of the four studies. In studies directly comparing 50 mg per day and 100 mg per day there was no suggestion of a greater effect with the higher dose and adverse reactions and discontinuations were more frequent at higher doses [see Dosage and Administration (2.1)].
| PRISTIQ | ||||||
|---|---|---|---|---|---|---|
| Study No. | Primary Endpoint: HAM-D17 | Placebo | 50 mg/day | 100 mg/day | 200 mg/day | 400 mg/day |
1 | Baseline Score (SD*) | 23.1 (2.5) | 23.2 (2.5) | 22.9 (2.4) | 23.0 (2.2) | |
Difference from Placebo (95% CI†) | -2.9‡ | -2.0 | -3.1‡ | |||
2 | Baseline Score (SD*) | 25.3 (3.3) | 24.8 (2.9) | 25.2 (3.2) | ||
Difference from Placebo (95% CI†) | -3.3‡ | -2.8‡ | ||||
3 | Baseline Score (SD*) | 23.0 (2.6) | 23.4 (2.6) | 23.4 (2.6) | ||
Difference from Placebo (95% CI†) | -1.9‡ | -1.5 | ||||
4 | Baseline Score (SD*) | 24.3 (2.6) | 24.3 (2.4) | 24.4 (2.7) | ||
Difference from Placebo (95% CI†) | -2.5‡ | -3.0‡ | ||||
Analyses of the relationships between treatment outcome and age and treatment outcome and gender did not suggest any differential responsiveness on the basis of these patient characteristics. There was insufficient information to determine the effect of race on outcome in these studies.
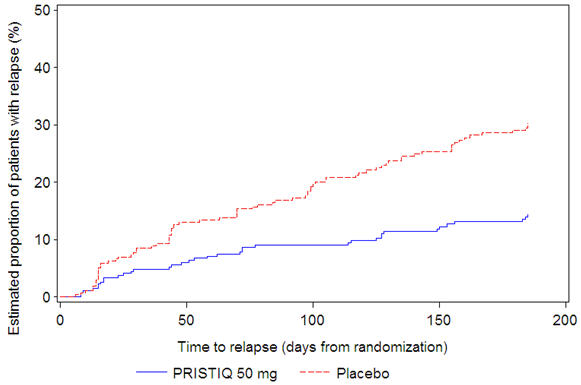
In a longer-term trial (Study 5), adult outpatients meeting DSM-IV criteria for major depressive disorder, who responded to 8 weeks of open-label acute treatment with 50 mg per day desvenlafaxine and subsequently remained stable for 12 weeks on desvenlafaxine, were assigned randomly in a double-blind manner to remain on active treatment or switch to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D17 total score of ≤ 11 and CGI-I ≤ 2 at the day 56 evaluation; stability was defined as HAM-D17 total score of ≤ 11 and CGI-I ≤ 2 at week 20 and not having a HAM-D17 total score of ≥ 16 or a CGI-I score ≥ 4 at any office visit. Relapse during the double-blind phase was defined as follows: (1) a HAM-D17 total score of ≥ 16 at any office visit, (2) discontinuation for unsatisfactory efficacy response, (3) hospitalized for depression, (4) suicide attempt, or (5) suicide. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse compared with placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 14% with desvenlafaxine treatment versus 30% with placebo.
Figure 4. Estimated Proportion of Relapses vs. Number of Days since Randomization (Study 5) |
 |
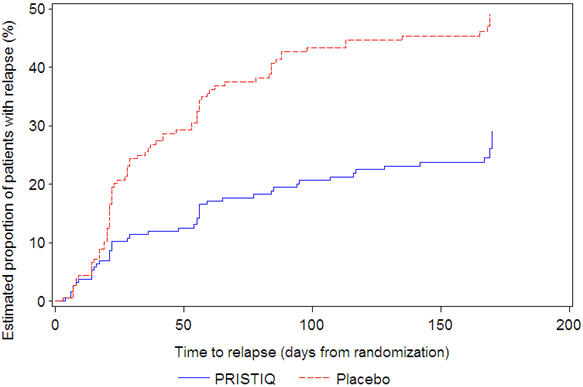
In another longer-term trial (Study 6), adult outpatients meeting DSM-IV criteria for major depressive disorder and who responded to 12 weeks of acute treatment with desvenlafaxine were assigned randomly to the same dose (200 or 400 mg per day) they had received during acute treatment or to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D17 total score of ≤ 11 at the day 84 evaluation. Relapse during the double-blind phase was defined as follows: (1) a HAM-D17 total score of ≥ 16 at any office visit, (2) a CGI-I score of ≥ 6 (versus day 84) at any office visit, or (3) discontinuation from the trial due to unsatisfactory response. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse over the subsequent 26 weeks compared with those receiving placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 29% with desvenlafaxine treatment versus 49% with placebo.
Figure 5. Estimated Proportion of Relapses vs. Number of Days since Randomization (Study 6) |
 |
In a postmarketing study, the efficacy of PRISTIQ at a dose lower than 50 mg per day was evaluated in an 8-week, multicenter, randomized, double-blind, placebo-controlled, fixed-dose study in adult outpatients with Major Depressive Disorder. The treatment arms were 25 mg (n=232), 50 mg (n=236), and placebo (n=231). The 50 mg dose was superior to placebo, as measured by the mean change from baseline on the HAMD-17. The 25 mg dose was not superior to placebo.
Find PRISTIQ® medical information:
Find PRISTIQ® medical information:
PRISTIQ® Quick Finder
Clinical Studies
14 CLINICAL STUDIES
Major Depressive Disorder
The efficacy of PRISTIQ as a treatment for depression was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose studies (at doses of 50 mg per day to 400 mg per day) in adult outpatients who met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for major depressive disorder. In the first study, patients received 100 mg (n = 114), 200 mg (n = 116), or 400 mg (n = 113) of PRISTIQ once daily, or placebo (n = 118). In a second study, patients received either 200 mg (n = 121) or 400 mg (n = 124) of PRISTIQ once daily, or placebo (n = 124). In two additional studies, patients received 50 mg (n = 150 and n = 164) or 100 mg (n = 147 and n = 158) of PRISTIQ once daily, or placebo (n = 150 and n = 161).
PRISTIQ showed superiority over placebo as measured by improvement in the 17-item Hamilton Rating Scale for Depression (HAM-D17) total score in four studies and overall improvement, as measured by the Clinical Global Impressions Scale - Improvement (CGI-I), in three of the four studies. In studies directly comparing 50 mg per day and 100 mg per day there was no suggestion of a greater effect with the higher dose and adverse reactions and discontinuations were more frequent at higher doses [see Dosage and Administration (2.1)].
| PRISTIQ | ||||||
|---|---|---|---|---|---|---|
| Study No. | Primary Endpoint: HAM-D17 | Placebo | 50 mg/day | 100 mg/day | 200 mg/day | 400 mg/day |
1 | Baseline Score (SD*) | 23.1 (2.5) | 23.2 (2.5) | 22.9 (2.4) | 23.0 (2.2) | |
Difference from Placebo (95% CI†) | -2.9‡ | -2.0 | -3.1‡ | |||
2 | Baseline Score (SD*) | 25.3 (3.3) | 24.8 (2.9) | 25.2 (3.2) | ||
Difference from Placebo (95% CI†) | -3.3‡ | -2.8‡ | ||||
3 | Baseline Score (SD*) | 23.0 (2.6) | 23.4 (2.6) | 23.4 (2.6) | ||
Difference from Placebo (95% CI†) | -1.9‡ | -1.5 | ||||
4 | Baseline Score (SD*) | 24.3 (2.6) | 24.3 (2.4) | 24.4 (2.7) | ||
Difference from Placebo (95% CI†) | -2.5‡ | -3.0‡ | ||||
Analyses of the relationships between treatment outcome and age and treatment outcome and gender did not suggest any differential responsiveness on the basis of these patient characteristics. There was insufficient information to determine the effect of race on outcome in these studies.
In a longer-term trial (Study 5), adult outpatients meeting DSM-IV criteria for major depressive disorder, who responded to 8 weeks of open-label acute treatment with 50 mg per day desvenlafaxine and subsequently remained stable for 12 weeks on desvenlafaxine, were assigned randomly in a double-blind manner to remain on active treatment or switch to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D17 total score of ≤ 11 and CGI-I ≤ 2 at the day 56 evaluation; stability was defined as HAM-D17 total score of ≤ 11 and CGI-I ≤ 2 at week 20 and not having a HAM-D17 total score of ≥ 16 or a CGI-I score ≥ 4 at any office visit. Relapse during the double-blind phase was defined as follows: (1) a HAM-D17 total score of ≥ 16 at any office visit, (2) discontinuation for unsatisfactory efficacy response, (3) hospitalized for depression, (4) suicide attempt, or (5) suicide. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse compared with placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 14% with desvenlafaxine treatment versus 30% with placebo.
In another longer-term trial (Study 6), adult outpatients meeting DSM-IV criteria for major depressive disorder and who responded to 12 weeks of acute treatment with desvenlafaxine were assigned randomly to the same dose (200 or 400 mg per day) they had received during acute treatment or to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D17 total score of ≤ 11 at the day 84 evaluation. Relapse during the double-blind phase was defined as follows: (1) a HAM-D17 total score of ≥ 16 at any office visit, (2) a CGI-I score of ≥ 6 (versus day 84) at any office visit, or (3) discontinuation from the trial due to unsatisfactory response. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse over the subsequent 26 weeks compared with those receiving placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 29% with desvenlafaxine treatment versus 49% with placebo.
In a postmarketing study, the efficacy of PRISTIQ at a dose lower than 50 mg per day was evaluated in an 8-week, multicenter, randomized, double-blind, placebo-controlled, fixed-dose study in adult outpatients with Major Depressive Disorder. The treatment arms were 25 mg (n=232), 50 mg (n=236), and placebo (n=231). The 50 mg dose was superior to placebo, as measured by the mean change from baseline on the HAMD-17. The 25 mg dose was not superior to placebo.
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.