TIVDAK® Clinical Studies
(tisotumab vedotin-tftv)
14 CLINICAL STUDIES
14.1 Recurrent or Metastatic Cervical Cancer
innovaTV 301
The efficacy of TIVDAK was evaluated in innovaTV 301 (NCT04697628), an open-label, active-controlled, multicenter, randomized trial that enrolled 502 patients with recurrent or metastatic cervical cancer who had received one or two prior systemic therapy regimens in the recurrent or metastatic setting, including chemotherapy with or without bevacizumab and/or an anti-PD-(L)1 agent. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy, or clinically significant bleeding issues or risks.
Patients were randomized (1:1) to receive either TIVDAK 2 mg/kg intravenously every 3 weeks (n=253) or investigator’s choice of chemotherapy (n=249) consisting of topotecan, vinorelbine, gemcitabine, irinotecan or pemetrexed, until unacceptable toxicity or disease progression. Randomization was stratified by prior treatment with bevacizumab (yes or no), prior anti-PD-(L)1 therapy (yes or no), region (US, Europe, or Other), and ECOG performance status (0 or 1).
Patients were treated until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were progression free survival (PFS) and confirmed objective response rate (ORR) as assessed by investigator using RECIST v1.1.
The median age was 50 years (range: 26 to 80); 49% were White, 36% were Asian, 10% were not reported, 3% were American Indian or Alaskan Native, and 2% were Black; 20% were Hispanic/Latino; and baseline ECOG performance status was 0 (54%) or 1 (46%). Sixty-three percent of patients had squamous cell carcinoma, 32% had adenocarcinoma, and 5% had adenosquamous histology. Ninety percent of patients had extrapelvic disease; 61% of patients had received 1 prior line of systemic therapy, and 38% had 2 prior lines of systemic therapy. All patients received prior chemotherapy; 64% received prior bevacizumab and 27% received prior anti-PD-1 or anti-PD-L1 therapy. Patients on the control arm received gemcitabine (44%), pemetrexed (32%), topotecan (8%), vinorelbine (7%), or irinotecan (6%).
Statistically significant improvements in OS, PFS, and ORR were demonstrated for TIVDAK compared with chemotherapy.
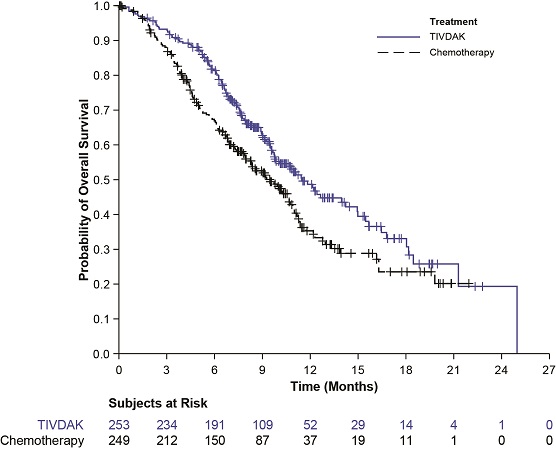
Table 9 and Figure 2 summarize the efficacy results from innovaTV 301.
| CI: confidence interval | ||
Endpoint | TIVDAK | Chemotherapy |
Overall Survival | ||
Number (%) of patients with events | 123 (48.6) | 140 (56.2) |
Median in months (95% CI) | 11.5 (9.8, 14.9) | 9.5 (7.9, 10.7) |
Hazard ratio (95% CI) | 0.70 (0.54, 0.89) | |
p-value | 0.0038* | |
Progression Free Survival | ||
Number (%) of patients with events | 198 (78.3) | 194 (77.9) |
Median in months (95% CI) | 4.2 (4.0, 4.4) | 2.9 (2.6, 3.1) |
Hazard ratio (95% CI) | 0.67 (0.54, 0.82) | |
p-value | <0.0001† | |
Confirmed Objective Response Rate (CR + PR) | ||
ORR (%) (95% CI) | 17.8 (13.3, 23.1) | 5.2 (2.8, 8.8) |
p-value | <0.0001‡ | |
Complete response rate (%) | 2.4 | 0 |
Partial response rate (%) | 15.4 | 5.2 |
Figure 2. Kaplan Meier Plot of Overall Survival
innovaTV 204
The efficacy of TIVDAK was evaluated in innovaTV 204 (NCT03438396), an open-label, multicenter, single-arm trial that treated 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy or known coagulation defects leading to an increased risk of bleeding.
Patients received TIVDAK 2 mg/kg intravenously every 3 weeks until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The median age was 50 years (range: 31 to 78); 95% were White, 2% were Asian, and 1% were Black. Six percent of patients were Hispanic or Latino. Sixty-eight percent of patients had squamous cell carcinoma, 27% had adenocarcinoma, and 5% had adenosquamous histology. ECOG performance status was 0 (58%) or 1 (42%). Seventy percent of patients had received 1 prior line of systemic therapy, and 30% had 2 prior lines of systemic therapy. Sixty-nine percent of patients previously received bevacizumab as part of their prior systemic therapy. Sixty-three percent received bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or carboplatin, or paclitaxel and topotecan) as first-line therapy.
The major efficacy outcome measures were confirmed objective response rate (ORR) as assessed by an independent review committee (IRC) using RECIST v1.1 criteria and duration of response (DOR).
Efficacy results are presented in Table 10.
| CI: confidence interval NR: not reached | |
| |
Endpoint | N=101 |
Confirmed ORR | 24% |
Complete response rate | 7% |
Partial response rate | 17% |
Duration of Response |
|
Median Duration of Response, months* | 8.3 |
Find TIVDAK® medical information:
Find TIVDAK® medical information:
TIVDAK® Quick Finder
Clinical Studies
14 CLINICAL STUDIES
14.1 Recurrent or Metastatic Cervical Cancer
innovaTV 301
The efficacy of TIVDAK was evaluated in innovaTV 301 (NCT04697628), an open-label, active-controlled, multicenter, randomized trial that enrolled 502 patients with recurrent or metastatic cervical cancer who had received one or two prior systemic therapy regimens in the recurrent or metastatic setting, including chemotherapy with or without bevacizumab and/or an anti-PD-(L)1 agent. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy, or clinically significant bleeding issues or risks.
Patients were randomized (1:1) to receive either TIVDAK 2 mg/kg intravenously every 3 weeks (n=253) or investigator’s choice of chemotherapy (n=249) consisting of topotecan, vinorelbine, gemcitabine, irinotecan or pemetrexed, until unacceptable toxicity or disease progression. Randomization was stratified by prior treatment with bevacizumab (yes or no), prior anti-PD-(L)1 therapy (yes or no), region (US, Europe, or Other), and ECOG performance status (0 or 1).
Patients were treated until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were progression free survival (PFS) and confirmed objective response rate (ORR) as assessed by investigator using RECIST v1.1.
The median age was 50 years (range: 26 to 80); 49% were White, 36% were Asian, 10% were not reported, 3% were American Indian or Alaskan Native, and 2% were Black; 20% were Hispanic/Latino; and baseline ECOG performance status was 0 (54%) or 1 (46%). Sixty-three percent of patients had squamous cell carcinoma, 32% had adenocarcinoma, and 5% had adenosquamous histology. Ninety percent of patients had extrapelvic disease; 61% of patients had received 1 prior line of systemic therapy, and 38% had 2 prior lines of systemic therapy. All patients received prior chemotherapy; 64% received prior bevacizumab and 27% received prior anti-PD-1 or anti-PD-L1 therapy. Patients on the control arm received gemcitabine (44%), pemetrexed (32%), topotecan (8%), vinorelbine (7%), or irinotecan (6%).
Statistically significant improvements in OS, PFS, and ORR were demonstrated for TIVDAK compared with chemotherapy.
Table 9 and Figure 2 summarize the efficacy results from innovaTV 301.
| CI: confidence interval | ||
Endpoint | TIVDAK | Chemotherapy |
Overall Survival | ||
Number (%) of patients with events | 123 (48.6) | 140 (56.2) |
Median in months (95% CI) | 11.5 (9.8, 14.9) | 9.5 (7.9, 10.7) |
Hazard ratio (95% CI) | 0.70 (0.54, 0.89) | |
p-value | 0.0038* | |
Progression Free Survival | ||
Number (%) of patients with events | 198 (78.3) | 194 (77.9) |
Median in months (95% CI) | 4.2 (4.0, 4.4) | 2.9 (2.6, 3.1) |
Hazard ratio (95% CI) | 0.67 (0.54, 0.82) | |
p-value | <0.0001† | |
Confirmed Objective Response Rate (CR + PR) | ||
ORR (%) (95% CI) | 17.8 (13.3, 23.1) | 5.2 (2.8, 8.8) |
p-value | <0.0001‡ | |
Complete response rate (%) | 2.4 | 0 |
Partial response rate (%) | 15.4 | 5.2 |
Figure 2. Kaplan Meier Plot of Overall Survival
innovaTV 204
The efficacy of TIVDAK was evaluated in innovaTV 204 (NCT03438396), an open-label, multicenter, single-arm trial that treated 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy or known coagulation defects leading to an increased risk of bleeding.
Patients received TIVDAK 2 mg/kg intravenously every 3 weeks until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The median age was 50 years (range: 31 to 78); 95% were White, 2% were Asian, and 1% were Black. Six percent of patients were Hispanic or Latino. Sixty-eight percent of patients had squamous cell carcinoma, 27% had adenocarcinoma, and 5% had adenosquamous histology. ECOG performance status was 0 (58%) or 1 (42%). Seventy percent of patients had received 1 prior line of systemic therapy, and 30% had 2 prior lines of systemic therapy. Sixty-nine percent of patients previously received bevacizumab as part of their prior systemic therapy. Sixty-three percent received bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or carboplatin, or paclitaxel and topotecan) as first-line therapy.
The major efficacy outcome measures were confirmed objective response rate (ORR) as assessed by an independent review committee (IRC) using RECIST v1.1 criteria and duration of response (DOR).
Efficacy results are presented in Table 10.
| CI: confidence interval NR: not reached | |
| |
Endpoint | N=101 |
Confirmed ORR | 24% |
Complete response rate | 7% |
Partial response rate | 17% |
Duration of Response |
|
Median Duration of Response, months* | 8.3 |
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.